D 5.4 Deliverable project month: Report on the result obtained
R. Beltrami, D. Medini, C. Donati, N. Pacchiani, A. Muzzi, A. Covacci
IRIS Bioinformatic Unit, Chiron S.p.A.
biocomp_admin_siena@chiron.it
1. Introduction
2. DNC-Blast
2.1 Avoiding databank reload from disks
2.2 Database randomization and splitting on non-homogeneous cluster nodes
2.3 Results (I)
2.4 Results (II)
2.5 DNC-Blast typical usage scenarios
2.6 Results for a real-case test
3 GROMACS
3.1 Configuration of a dedicated "run-level" for MD tasks
3.2 Network communications: Fast-Ethernet vs. Gigabit-Ethernet
4 Conclusions
5 Acknowledgements
6 Appendix A
1. Introduction Top
Several successful experimentsi have already been done on the
parallelization of commonly used bioinformatic tools. The scope of this
project was to tailor standard algorithms widely used in genetic data
analyses, the NCBI Blast and GROMACS, for parallel architectures and
evaluate performances into our "in house"lf/sito/ facility. The project
was segmented into two phases: i) development of a prototype and ii)
its implementation. In the first phase the work was done on a Beowulf
cluster while the second phase was completed using a Production
Facility (PF) of industrial strength.
Maximum portability and usability was achieved using i) Unix
multiprocessor servers and ii) cluster of workstations with software
codes and customization done using standard and freely available
software tools.
In this workpackage we have tuned and optimized the DNC-Blast engine -
the BioWulf's parallelized version on the NCBI-Blast - on the PF
(cluster and SMP). We have tried to i) minimize performance degradation
caused by reloads of data from the storage subsystem; ii) achieve a
better load-balancing by randomization and splitting of the databases
on the PF cluster. Than we have tested system performances under heavy
load and we have compared, in a "real-case"lf/sito/ test, the newly
developed tool (DNC-Blast) with the original one (NCBI-Blast). We have
also completed the optimization of Gromacs on the PF cluster, and we
have compared the performances of two different network communication
systems.
The collected data suggest that the Production Facility (cluster and
SMP) is particularly well suited for massive data analysis and that,
with the developed tools, it is possible to greatly improve the system
performances, as required from high-end genomic research projects such
as grouping of protein families.
2 DNC-Blast
2.1 Avoiding databank reload from disks Top
We have already showni that the main part of the overall
execution time of a Blast query was the I/O reload time of the
databank(s) from the disk subsystem into the main memory. Therefore a
strategy to minimize the I/O time was required. One possible solution
is the implementation and use of a RAM-disk (i.e. a virtual disk driver
that mimics a disk subsystem using the main memory as storage space).
Sun Solaris (release 2.5.1 and above) provides a similar device, namely
the Temporary File-System tmpfs, that uses the RAM memory and the swap
disk space as storage area and is mounted under the /tmp mount point by
default. However, the tmpfs actually does not implement a pure RAM-disk
since data stored in /tmp could be swapped out to let other data use
the RAM clearly diminishing the performances.
Another possibility to allocate the databank in main memory is through
the operating system call mlock(3C) and related callsii. This function
can permanently force (until a subsequent call to munlock(3C)) a
defined area of the process virtual memory to reside in system main
memory avoiding I/O delays. However two major drawbacks come out with
this solution: firstly, one need to directly modify the NCBI-Blast
source files in order to use the mlock function; secondly, the mlock
function can be used only by processes running with administrator
privileges, thus adding potential system stability and security
concerns.
Since the development of a custom, platform-specific, high-performance
RAM-disk driver or the modification of the NCBI-Blast internal
structure both fall out of the scopes of the present project, we have
fine-tuned the tmpfs configuration to minimize databank reload times.
Actually, with such a configuration, we have not been able to measure
any delay in our internal usage of the DNC-Blast due to databank
reloads.
2.2 Database randomization and splitting on non-homogeneous cluster nodes Top
As
reported in a previous deliverablei, in order to setup the DNC-Blast
environment, both on the PF SMP and on the PF cluster, every databank
being searched needs to be partitioned. The original NCBI Blast suite
includes a tool for formatting -creating indexes- on the source textual
data files. This program, namely formatdb, also allows to split the
source file into one or more indexed files. Originally this was
developed to circumvent the maximum file size limit imposed by older
32-bit operating systems (with a maximum file size of 2GB). Splitting
the database to be searched when using the original multithreaded
NCBI-Blast on a single SMP machine has no impact whatsoever on the
performances. Indeed, this feature could be used to generate the
databank slices as previously mentioned4. This procedure has two
drawbacks. A: the splitting method provided by formatdb simply puts the
first part of the input source file into the first resulting slice, the
second into the second slice (and so on); B: the slices will result of
the same size. Two major consequences will occur: when part of
databanks are searched by the DNC-Blast sub-processes, it is most
likely that an uneven distribution of "sequence types"lf/sito/ will be
accessed and an unbalanced load will result. Since the databank source
files often contain sequences from the same organism clustered together
and similar organism sequences close one to each other, groups of
similar sequences will be probably contained in the same slice and
slices produced by formatdb will be biased on the type of organism they
belong to. As an example, if we search a databank using a query
sequence from a bacterial organism, the slices containing the bacterial
sequences will show far more hits (i.e. longer execution time for HSP
searching and alignment extension) than those containing data from
other organisms and the overall execution time will depend on them. In
addition, given the heterogeneous nature of the PF cluster, to improve
the load balancing among the nodes it's mandatory to fragment the
databanks in slices proportional to the computational power of each
single node.
We have solved the first issue by randomly changing the order of the
input sequences to obtain a uniform distribution of all the "sequence
types"lf/sito/ along the whole databank. Then applying the original
formatdb we were able to obtain non-biased slices of equal size. These
slices were used to improve the performances of DNC-Blast on the PF
SMP, and the results are reported in the Results(I) section.
We also have implemented a custom version of the NCBI formatting
program, the DNC-formatdb, able to generate randomized database slices
of variable sizes. The DNC-formatdb developed in this workpackage takes
as input the databank to be formatted and a set of relative weights
(expressed as percentual figures) of every slice to be generated. Then,
it produces randomized slices with sizes varying according to input
weights ready to be searched using DNC-Blast.
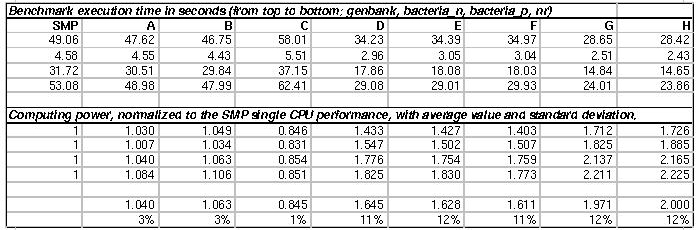
Table1
EqCPU numbers computed for each databank
To perform this task a modified randomizing algorithm has been applied.
For each single input sequence a random number between 0 and 1 has been
generated and the sequence assigned to one of the output file depending
on the numeric range the number falls in. The slices obtained so far
are then simply fed into the NCBI-formatdb to generate five searchable
files to be distributed on the computing nodes. In practice, to
distribute each databank over the nodes we have implemented the
following procedure:
compute the execution time of a reference Blast query (one for each
databank) on a reference machine, namely a single CPU of the PF SMP.
This number, from now on called one Equivalent CPU (1 EqCPU),
represents the measure unit of the execution times of the reference
query on the cluster nodes;
measure the execution time of the reference Blast query (one for each
databank) on each node and compute the EqCPU value for the node as the
ratio with the execution time on the reference machine;
compute the relative weight of the databank slice for each node (one
for each databank) based on the number of EqCPU of that node;
run the DNC-formatdb to generate the randomized and weighted databank
slices to be distributed on each node.
Table 1 shows the execution times and the computed EqCPU values, for
each databank, on all the cluster nodes. The database slices obtained
(whose weights are reported in Appendix A) were used to improve the
performances of the DNC-Blast on the PF-cluster, and the outcomes are
reported in the Results(II) section.
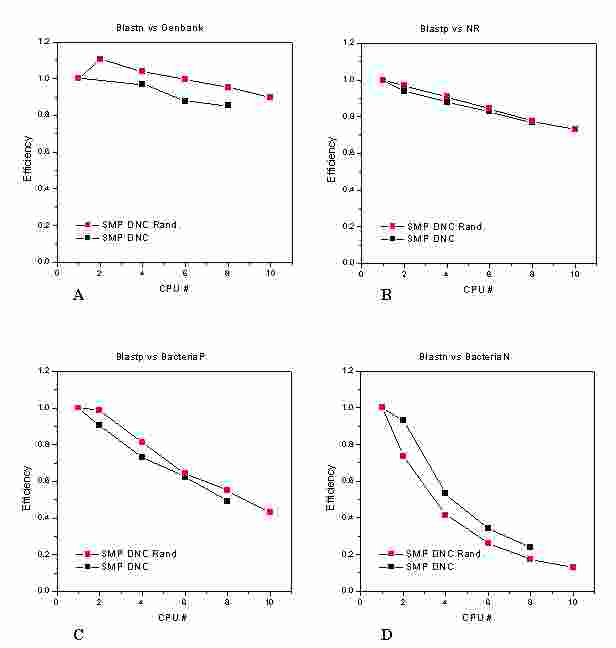
Figure 1
A efficiency increase close to 10% for queries
against the randomized GenBank is obtained, for the other databanks the
change is less pronounced, and negative in the BacteriaN case
2.3 Results (I)
Top
Tests conducted to evaluate the effectiveness of the database
randomization on the PF SMP. We have repeated the tests performed in a
previous deliverablei, for BlastN and BlastP against four different
databases, both on non-randomized databank slices and on randomized
ones. In all cases the same DNC-Blast implementation was used, being
the database randomization the only difference among the two tests. In
Figure 1 the efficiencies of the algorithm are shown as a function of
the CPU number n for the randomized (red points) and non-randomized
(black points) databases: "A" for BlastN against the Genbank, "B" for
the BlastP against the NR, "C" for BlastN against Bacteria_P and
"D"lf/sito/ for Bacteria_N. The efficiency is defined as the ratio of
the theoretical minimum execution time with n processors and the actual
elapsed timeii, where the theoretical minimum execution time is given
by the execution time of one processor divided by the number of
processors n.
Results show a performance increase due to the randomization procedure
for three of the four databases under consideration. In particular, for
BlastN against Genbank the performance exceeds the theoretical value at
2 and 4 CPUs, showing a behavior similar to a well known
cache-aggregate effect, made possible in the randomized case for the
high degree of balance among the concurrent sub-processes. The
efficiency gain is less evident with BlastP ("B" and "C"), where the
more complex structure of the alignment algorithm for protein sequences
reduces the impact of the data parallelization. When small databases
are searched with a fast algorithm (as for BlastN against Bacteria_N in
"D") we register a reduced efficiency in the randomized case.
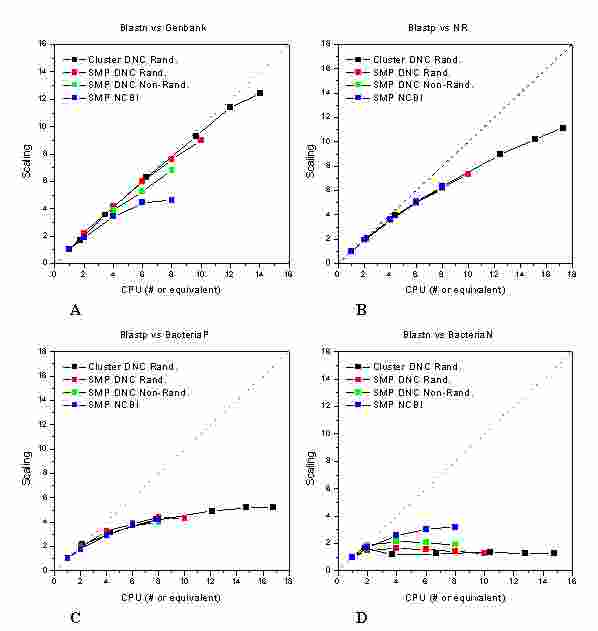
2.4 Results (II)
Top
Tests
on heterogeneous database splitting on the PF cluster
Following the procedure outlined in the previous section, we have
populated the cluster with heterogeneous randomized databank slices. In
the scalability of the algorithm (defined in our previous testi) is
shown as a function of the EqCPU number in order to provide a correct
comparison among the two platforms. It should be reminded that the
EqCPU for the cluster nodes are defined using one single CPU of the PF
SMP as a unitary reference. Hence, for the PF SMP one EqCPU is exactly
one processor while, for the PF cluster, the EqCPU is neither an
integer nor it corresponds to the number of the nodes.
In each graph, four different data sets are shown:
the multithreaded NCBI-Blast using databanks formatted with standard
formatdb as distributed in the NCBI toolkit (blue squares)
the DNC-Blast on the PF SMP using databanks from the standard formatdb
(green squares)
the DNC-Blast on the PF SMP using databanks from the customized
DNC-formatdb, in this case all the databank slices are equal in size
(red squares)
the DNC-Blast on the PF cluster using databanks from the customized
DNC-formatdb using slice sizes as previously reported (black squares)
In the Genbank graph ("A"lf/sito/) the DNC-Blast algorithm
over-performs the NCBI-Blast on the PF SMP of a factor 1.6, 1.9 in the
randomized case, and the heterogeneous splitting of the database allows
to obtain an excellent performance also on the PF cluster. The PF
cluster performs 2.6 times better than the PF SMP with the same number
(8) of actual processors, being some of the CPU of the cluster in this
experiment more powerful than a single SMP processor.
For the protein databanks (NR in "D" and BacteriaP in "C"lf/sito/) we
don't have any relevant performance gain due to the DNC-Blast algorithm
itself. We notice that the BlastP algorithm have to deal with a much
more complex alphabet than BlastN, which results in a much longer time
spent in the HSP expansion phase. Being this the already parallelized
portion of the NCBI-Blast code, the DNC-Blast data-parallelization
strategy results in a less marked performance gain on the PF SMP
platform. Nevertheless, as in the Genbank case, it allows to use the
cluster platform with the same good results, particularly with the NR
database where a performance ratio of 11/7 is visible when all the CPUs
are in use. Conversely, for the BacteriaN ("D"lf/sito/), we observe a
severe degradation of the performances on the cluster, showing
Figure 2
Scalability of DNC-Blast (PF SMP and cluster) vs. NCBI-Blast (PF SMP).
that with fast queries on small databases the data collection times
become extremely relevant in the overall execution time of the process.
2.5 DNC-Blast typical usage scenarios
Top
In
order to improve the overall performance of the Production Facility, we
have considered the real usage made for computing intensive genomic
research. The typical usage of a Blast engine can be divided into two
broad categories: interactive and batch execution. The first is implied
whenever a user performs a Blast search for a single (or a small set
of) query sequences against one databank. The second is typically made
of a huge set of query sequences sequentially searched against one or
more databanks and is usually run through a shell script that iterates
the task.
The two categories imply different resource usage schemes: the first is
mainly interactive and the loading order of databanks is not
predictable; in the second all the search jobs are known in advance and
can be arranged to better exploit the platform performances.
In the first case the PF SMP is more suited to correspond to the user
requests, given its much higher flexibility in dynamically assigning
resources to the tasks that change continuously; here the DNC-Blast can
be useful, as we have seen in the previous section, when big nucleotide
databases are searched. Conversely, the DNC-Blast algorithm can make a
real difference in the second case, when the user requests are much
more predictable, and the static allocation of the resources typical of
the BioWulf cluster is not an issue. Hence it can be of great
importance for computational intensive research projects such as
protein family classification, assigning gene functions, etc. In the
next paragraph we report the results of a test that mimics these tasks.
In this "real-case" use of the DNC-Blasti on the PF cluster we have
considered the concurrent usage of different query sequences and
databanks to effectively measure actual execution times.
2.6 Results for a real-case test
Top
It
is well established protocol that when a new complete bacterial genome
sequence is published or draft versions become available a complete
Blast search against public and in-house databanks is immediately
carried out. In fact, Blast results are the basis for further analysis,
for example for protein family classification or for genome annotation
or comparative genomics.
Here we report the results of a "real-case"lf/sito/ test where we have
compared the behavior of the DNC-Blast on the PF cluster with
fine-tuned tmpfs and the randomized heterogeneous database slices and
of the original NCBI-Blast on the PF SMP. We used the complete genome
sequence of Streptococcus pneumoniae strain R6 as released on GenBanki.
We took all the predicted genes and the encoded protein sequences and
DNC-blasted against three of the four DBs used in this project. A
whole-genome Blast search (both when the predicted genes or the encoded
proteins are used as input queries) consists of a series of Blast
commands (blastP or blastN accordingly) run sequentially, each
producing a single output file and finally collecting the Blast reports
of all the homology searches. The case of a search against a single
databank is far from the real use since more than a databank is scanned
at the same time, requiring balanced distribution of the load on the
nodes. Hence, to give a reasonable estimate of the real activity
performed by our Blast-engine, we have concurrently run three different
whole-genome Blast searches against the Genbank, the Non Redundant
protein database and the in-house bacterial genome database Bacteria_N.
The three sequential multi-input Blast where run concurrently, using
the DNC-Blast on the PF cluster and the NCBI-Blast on the PF SMP for
comparison. First of all, we have estimated the execution time of the
three jobs on the two platforms in order to achieve a balanced load
that minimizes the overall execution time. For the PF SMP, on a trial
and error base, 8 CPUs where assigned to the Genbank search, 2 CPUs to
the NR and one CPU to the Bacteria_N. This showed to be the best
balance achievable in order to minimize the overall execution time. The
remaining CPU (from a total of 12) was left free for system needs. On
the PF cluster, on a trial and error basis, 5 CPUs where assigned to
the Genbank job, 4 to the NR and one to the Bacteria_N.
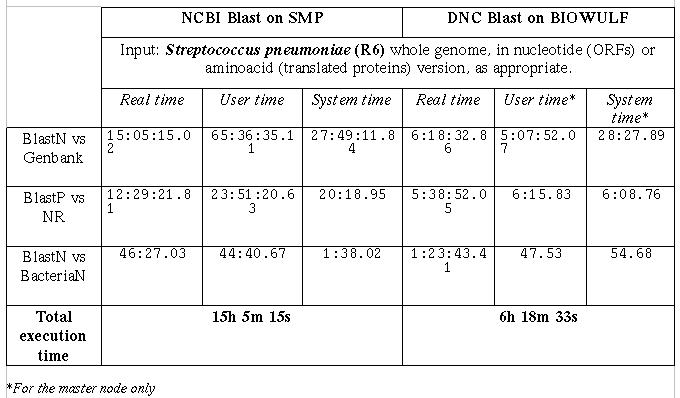
Table2
Times required for a complete genome search against three databases
Table 2 shows the timing results for the two platforms under
comparison. The databanks are the same used in the previous test. The
Real time columns is the relevant one for the evaluation of the overall
performances. The Total execution time is the maximum of real execution
times of the three concurrent processes.
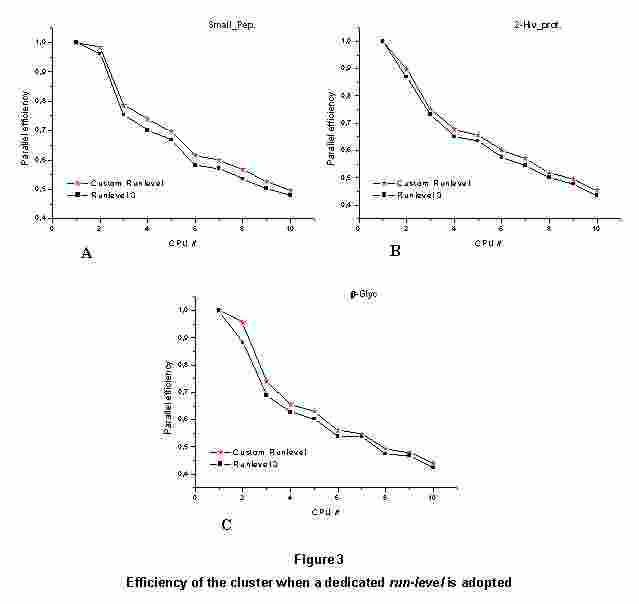
3.1 Configuration of a dedicated "run-level" for MD tasks
Top
In previous workpackagesi,ii we have reported how the molecular
dynamics (MD) program Gromacs have been implemented on the prototype
Beowulf cluster, and then ported on the PF cluster. We have also
shown11 that with an accurate fine tuning of the MPI implementation and
of some system parameter on the PF cluster it is possible to improve
somehow the rather poor parallel scaling of the MD algorithm. In this
section we report the results of a further attempt to increase the
parallel performances of an MD simulation on a cluster of workstations
(such as the PF cluster). Using workstations not specifically dedicated
to the cluster has the downside of the presence of many tasks and
services running on each workstation that consume system resources. In
fact, most of the nodes have common daemons running sendmail, the X
windows server, http server, etc. that diminish the performances of
parallel jobs.
Since molecular dynamic simulations are typical batch jobs and can be
run off-time (i.e. when the workstations are not used for other
purposes) one possible way to improve performances is to shut down all
the system processes and daemons not strictly required to run the
cluster. More precisely, the services required for the cluster are: the
NFS client to access data and executable files on the master node and
the daemons required for cluster communications (MPI layer), plus the
very basic system processes to keep the operating system running.
This running state can be defined by means of a custom run-level
operated through the usage of the command init. We defined a custom
run-level that is used for MD simulations.
Figure 3 shows the results obtained running the three usual MD tests
(already defined in a previous workpackage11) in the specific run-level
compared with those in the default run-level. As expected, we can
appreciate only a small increase (about the 5%) in the performance. The
small gain remains almost constant when increasing the CPUs number,
thus becoming relevant above 8 CPUs. In fact, after that number, the
introduction of the custom run-level gives a performance increase close
to that of adding another CPU to the cluster.
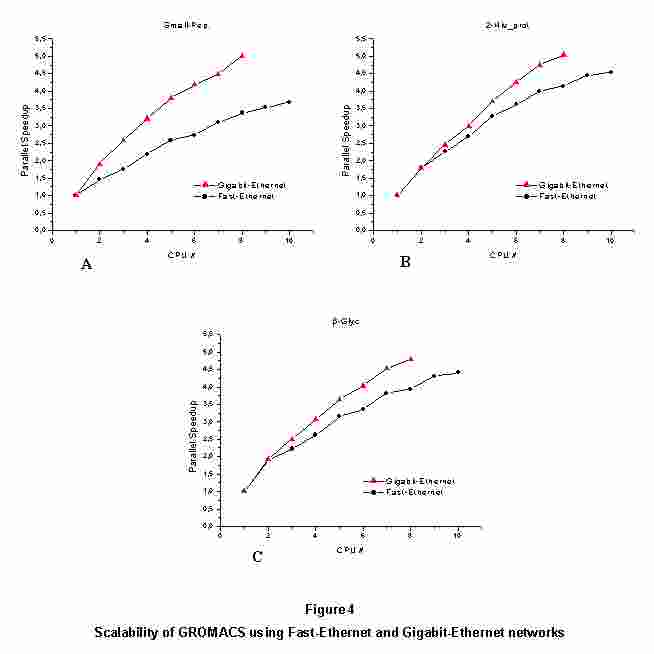
3.2 Network communications: Fast-Ethernet vs. Gigabit-Ethernet
Top
We have previously described10 how the high rate of communications
between nodes generated by the parallel algorithm implemented and the
type of network used limit the simulation time and the scalability of
this software package. To further explore how to improve the
performance of the PF cluster we conducted a series of tests using two
different network devices: Fast-Ethernet and Gigabit Ethernet. The
cluster using Fast-Ethernet network was composed of five bi-processor
workstation SunBlade1000 equipped with UltraSparc III CPU at 750MHz
while the cluster with Gigabit Ethernet was composed of four
bi-processor E280R equipped with UltraSparc III at 750MHz. Both
clusters were connected using Cisco network switches.
The results obtained from the simulation sets (see Figure 4) confirm
that the network layer is the limiting factor for performance and
scalability. The Gigabit network showed known limitations of Ethernet
networks for this kind of traffic and scalability is still far from
optimal. However, Gigabit cluster produced better results: using 8
processors the scalability is generally more than one unit above the
Fast-Ethernet network.
Gigabit connections can be seen as preferred over the older one: even
with a relatively small number of nodes the Gigabit cluster can give
the same or better simulation time using one or two processors less
than the Fast-Ethernet so the higher cost of the Gigabit network
interfaces is balanced by a lower cost of the workstations. In
addition, the availability of Gigabit Ethernet of copper wire (UTP
cables) instead of optical fibers further simplify the adoption of this
technology as a substitute of the older one.
4 Conclusions
Top
DNC-Blast
The Basic Local Alignment Sequence Tool (BLAST) is a typical example of
bioinformatic tool that deals with the exponentially growing amount of
genomics data available to the scientific community. Given the
intrinsic nature of a homology search algorithm, the wider the data set
the poorer the parallel efficiency of the algorithm itself. Starting
from this evidence we have tried to apply a different parallelization
strategy, namely the parallelization of the databank. The challenge was
to show that such a strategy can be successfully applied on traditional
architectures (such as our Production Facility Symmetrical
MultiProcessing) and also on lighter structures such as our Production
Facility cluster of heterogeneous workstations, here called BioWulf.
This without interfering with the internal structure of the NCBI-Blast
itself. We started by fractioning the databank using the indexing tool
of the NCBI-Toolkit, formatdb. Then, we developed a software tool,
called Divide aNd Conquer (DNC)-Blast, that performs a set of single
Blast queries on portions of the databank and merges the results in a
single output report. With an appropriate treatment of the statistical
inferences, this output is completely equivalent with the original one.
The next step was to face with the performances of such an algorithm,
beginning with the PF SMP architecture. As it could be expected, on
small data sets we found poor performance improvements while, a sharp
gain in the parallel efficiency have been detected when increasing the
database size, especially for nucleotide sequence alignments. Further
improvements have been achieved by randomizing the databanks to be
searched reaching, for BlastN queries against the Genbank, a
performance gain close to 2 times.
While porting the tool on the PF cluster, we found that the standard
NCBI-formatdb indexing tool was not able to properly handle the
heterogeneity of the BioWulf, then we developed a custom DNC-formatdb,
that splits the databanks in randomized weighted portions, highly
improving the balance of the computational load on the cluster nodes.
In this way we showed that it is possible to use an heterogeneous
cluster architecture, with any performance degradation with respect to
the SMP, with all the intrinsic advantages.
As a final result, we executed a typical task of a genomic research
project with both the NCBI-Blast and the DNC-Blast: the latter
completed in less than half the time of the former.
Gromacs
The GRoeningen MAchine for Computer Simulations (GROMACS) is a widely
used molecular simulation program, with an algorithmic structure
completely different from sequence alignment tools. As opposite to the
Blast, a parallel MPI version of the code, suited for a cluster
architecture, was already available before the start of the present
project. The main challenge here was to show if and how a PF cluster,
not specifically designed as a MD-engine, could perform the tasks. An
initial study on a prototype cluster of various MPI implementations
confirmed that network communications are the real bottleneck of the
process. Hence, when porting the package on the PF cluster, we tried to
fine-tune the configuration of the network and of the system itself,
obtaining a performance increase that allowed us to stay close, for
some simulated system, to the Amdhal's law limit. Finally we showed
that switching to the Gigabit-Ethernet, a faster yet still affordable
network connection, it was possible to achieve a performance gain of
1.5 times.
In conclusion, we showed that the algorithm used for
DNC-Blast is particularly well suited for parallel systems and can be
utilized with no modification on SMPs as well as on BioWulf clusters.
Also hybrid solutions that employ both architectures can be applied.
Present day biology requires to analyze an increasing amount of data in
as short time as possible. As DNC-Blast gave excellent results when
used on large volumes of data (while on small data sets it was almost
impossible to obtain better results than the original Blast because of
the low execution times already achieved) it showed to be a brilliant
yet industrial strength bioinformatic tool for genomics projects.
The production facility demonstrated to be a high performance platform
suitable to execute iterative tasks on data sets in the order of
several gigabyte in size. As an example, the grouping of protein
sequences in meaningful families requires the execution of
"all-to-all"lf/sito/ Blast alignments on a set of hundreds of thousands
sequencesi. This took up to thirty days of continuous elaboration:
properly increasing the number of nodes used by a DNC-Blast engine
would cut the computing time by an order of magnitude allowing a more
precise definition of the protein families in the same time.
Also, the results obtained suggested that the parallelization strategy
used for DNC-Blast can be profitably employed for other sequence
alignment tool such as the Smith&Waterman searchii. A similar
approach could be applied to this non-heuristic sequence alignment tool
- that provide more precise and sensible results than Blast -
overcoming the higher computing resources required by mean of a
parallel implementation.
5 Acknowledgements
Top
Part
of the present workpackage -more precisely the part on Gromacs testing-
has been completed with the helpful collaboration Carlo Nardone, Paolo
Carini and Alfredo Polizzi.
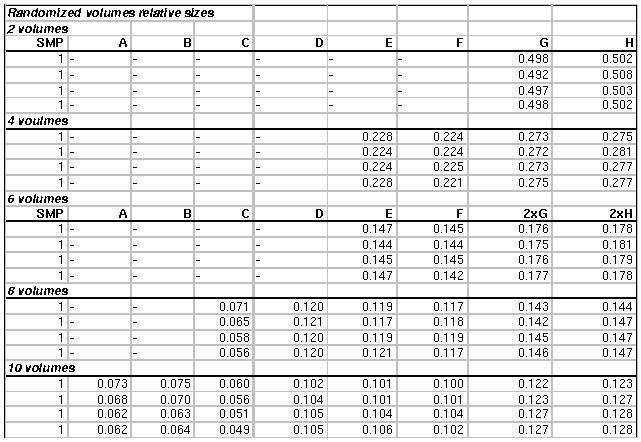
6 Appendix A
Top
Table3
Here we report
the database volume sizes obtained according to the Equivalent CPU
values listed in Table 1. Since the computing power slightly depends on
the application employed (blastp or blastn) and on the overall databank
size, the splitting is different for the four databanks. In Table 3,
from top to bottom, are listed the volume relative sizes for the
Genbank, the BacteriaN, the BacteriaP and the NR. After the
heterogeneous randomization of the database and indexing by mean of
formatdb, the single volumes have been moved locally on the assigned
nodes. It should be observed that all the nodes are used with a single
CPU per node, with the exception of the two Blade 1000@900 MHz, which
are used with all the two CPUs available per node in the 6,8 and 10
volumes decomposition.
HOMEPAGE